MLST
Multi-Locus Sequence Typing is an "approach that enables the unambiguous characterization of bacterial isolates in a standardized, reproducible, and portable manner" -NIH
About
MLST schemes are based around a community-agreed set of 7 gene loci present in all strains of the species. A database of validated allele sequences is maintained for each locus and a code assigned to each one. An "ST" code is then generated from the unique combination alleles. The schemes supported by Pathogenwatch are provided by PubMLST, while an in-house search tool is used to rapidly but accurately assign the correct MLST assignment.
Novel allele and MLST codes are indicated by the "*" character at the start of the code. It may also contain letters instead of numbers. Novel loci codes will be consistent between releases, unless replaced by an assignment from the host scheme.
If your profile includes novel alleles or a novel MLST code, we recommend visiting the source database linked in the results page to submit your genome there. Generated assignments will subsequently be imported in Pathogenwatch at the next update.
As described on PubMLST:
Multilocus sequence typing (MLST) is an unambiguous procedure for characterising isolates of bacterial species using the sequences of internal fragments of (usually) seven house-keeping genes. Approximately 450-500 bp internal fragments of each gene are used, as these can be accurately sequenced on both strands using an automated DNA sequencer. For each house-keeping gene, the different sequences present within a bacterial species are assigned as distinct alleles and, for each isolate, the alleles at each of the seven loci define the allelic profile or sequence type (ST).
Each isolate of a species is therefore unambiguously characterised by a series of seven integers which correspond to the alleles at the seven house-keeping loci.
...
The great advantage of MLST is that sequence data are unambiguous and the allelic profiles of isolates can easily be compared to those in a large central database via the Internet (in contrast to most typing procedures which involve comparing DNA fragment sizes on gels). Allelic profiles can also be obtained from clinical material by PCR amplification of the seven house-keeping loci directly from CSF or blood. Thus isolates can be precisely characterised even when they cannot be cultured from clinical material.
MLST profile construction
An MLST profile consists of a defined number of non-overlapping loci, usually seven. For each locus, a single code representing a specific allele is reported (an “ST” code), while the combination of alleles is also reported using a unique identifier. For each MLST scheme, a single provider maintains the allele and combined ST codes, and assigns a positive integer code for each unique entity. More recently identified alleles will have larger integer codes.
When searching a genome against a scheme, it is possible for there to be cross-hits or overlaps between potential locus matches, as well as duplications either from assembly error or genomic rearrangement. Firstly only a limited overlap is allowed between matches, typically less than 30 nucleotides. If there are multiple matches to different regions (e.g. paralogs) then exact matches to known alleles are selected in preference for the MLST profile. If there are multiple known alleles present, then the one with the oldest (lowest integer) ST code is selected as the representative. If there is no exact match then the matches are sorted according to match length and then sequence identity against the known alleles and the top hit chosen as the (novel) allele. If there are still two equally plausible matches the one close to the oldest allele is selected.
Method
The assembly is searched for exact matches to known alleles. A representative set of alleles for each locus are then searched for using Blast. These searches are combined and filtered based on the similarity of the match and length of the match. Novel alleles are hashed using the SHA-1 algorithm, this is then used as their unique identifier. Profiles are assigned based on the combination of alleles detected. Novel profiles are also given a unique identifier using the SHA-1 hash algorithm.
Results
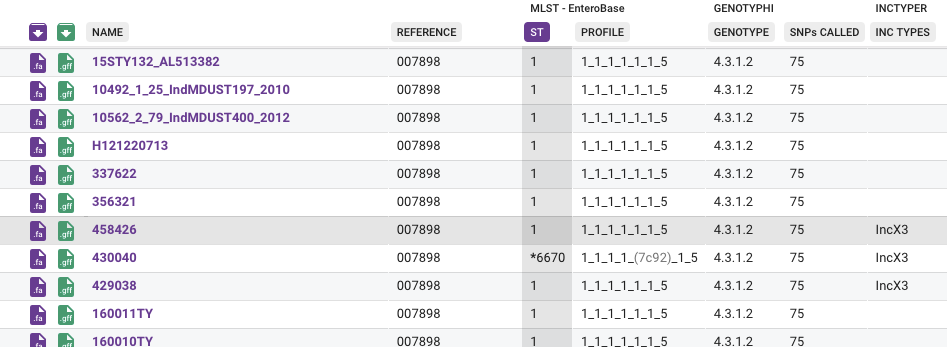
For each assembly the assigned allele codes and combined ST code is provided. If a locus is missing, the allele is represented with a question mark, while if it is a novel allele a four letter code that uniquely represents that allele is shown.
Alleles that begin with an asterix, such as *6670, are novel alleles and do not have an assigned numeric code. Novel MLST profile codes are reported in the same way. This appearance is different from some other MLST implementations. In the Collection View a novel ST due to a combination of alleles is shown as a unique four letter code, while those due to a new allele also have an asterisk ("*") marking them.
If a novel allele has been assigned, we advise against using it in a publication as it will be replaced once classified by the host resource.
"How it works" description
See https://github.com/pathogenwatch-oss/mlst?tab=readme-ov-file#how-it-works
How to cite
Please cite the resource which hosts the MLST scheme. The host of the scheme should linked in individual genome reports. Please contact us if you have any questions.
The software is available under an OSS licence from https://github.com/pathogenwatch-oss/mlst and https://github.com/pathogenwatch-oss/typing-databases.
References:
See https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3988353/
Code Repository
See https://github.com/pathogenwatch-oss/mlst and https://github.com/pathogenwatch-oss/typing-databases
Validation of MLST assignments for PATH-SAFE
Validation testing against EnteroBase showed a high concordance between the expected and assigned ST codes, suggesting the PATH-SAFE pipeline correctly places genomes in the correct groups. Furthermore, testing against the original assemblies showed 100% concordance with EnteroBase, giving confidence that the CGPS in-house tool is correct in general.
Validation report
The full validation report for PATH-SAFE can be found HERE.
Last updated